
El postulado de que la temperatura absoluta es directamente proporcional a la energía cinética promedio de las moléculas fue anticipado conceptualmente por Daniel Bernoulli en 1738.
Con este postulado Daniel Bernoulli se adelantó más de un siglo a la ciencia de su tiempo. Su propuesta apareció en la sección décima de su obra maestra de 1738, «Hydrodynamica» (cuyo manuscrito terminó de redactar en 1733, aunque se imprimió unos años después). [1, 2, 3]
Bernoulli propuso este modelo mecánico cuando la comunidad científica creía casi unánimemente que los gases estaban compuestos por átomos estáticos que se repelían a distancia, y que el calor era un fluido material.
En el siglo XVIII, la palabra «atomismo» como la entendemos hoy (la teoría de John Dalton de 1803 con elementos químicos fijos y pesos atómicos) no existía ni era aceptada. [1] Sin embargo, gracias a científicos del siglo XVII como Robert Boyle e Isaac Newton, casi toda la comunidad científica aceptaba la «teoría corpuscular». Esta teoría dictaba que la materia estaba hecha de partículas mecánicas diminutas llamadas «corpúsculos». [1, 2, 3, 4, 5]
En su obra cumbre, los Principia Mathematica (Libro II, Proposición 23) de 1687, Newton demostró matemáticamente que si se asumía que un gas estaba compuesto por partículas estáticas que se repelían mutuamente con una fuerza inversamente proporcional a la distancia entre sus centros, el resultado macroscópico explicaba a la perfección la Ley de Boyle 
Como Newton era considerado una autoridad casi indiscutible, la comunidad científica adoptó casi unánimemente su modelo estático. Imaginaban el gas como una esponja o un conjunto de resortes: los corpúsculos estaban quietos, flotando en el espacio, empujándose unos a otros a distancia mediante fuerzas repulsivas. [1, 2, 3]
A Bernoulli se le ocurrió una idea alternativa aplicando de forma directa las leyes de la mecánica de Newton al mundo microscópico, combinándolas con su intuición sobre los fluidos, que era distinta a la de Newton:
- La analogía del émbolo y los proyectiles: Bernoulli imaginó un cilindro vertical con un pistón móvil cargado con un peso. Se preguntó: ¿por qué el aire sostiene este peso y no se colapsa? En lugar de pensar en fuerzas repulsivas estáticas, imaginó que el espacio estaba lleno de miles de «corpúsculos diminutos» moviéndose a toda velocidad. El pistón se mantenía a flote por el bombardeo continuo e incesante de estas partículas.
- Geometría y proporcionalidad: Razonó que si comprimía el gas a la mitad de su volumen ($V/2$), habría el doble de partículas en el mismo espacio y chocarían el doble de veces contra el pistón. Así dedujo mecánicamente la Ley de Boyle (
), comprobando que su modelo matemático encajaba perfectamente con la realidad experimental de la época.
- La conexión con el calor de Amontons: Pocos años antes (1702), Guillaume Amontons había inventado un termómetro de aire y notó que la presión de un gas aumentaba al calentarse. Bernoulli unió los puntos: si la presión dependía del impacto de los corpúsculos y el calor aumentaba la presión, el calor debía ser el responsable de acelerar las partículas. [1, 3, 4, 5]
Bernoulli no utilizó el término moderno de «energía cinética» (que no se acuñaría hasta el siglo XIX), sino que habló de la «velocidad de los corpúsculos» y de la «fuerza viva» (vis viva, el precursor de la energía cinética, que en esa época se definía matemáticamente como 
«Los corpúsculos están dotados de un movimiento rapidísimo; […] el fluido elástico está compuesto de corpúsculos sumamente pequeños, que se mueven de un lado a otro con gran velocidad, y su presión se produce por los impactos de estos corpúsculos contra las paredes». [1]
Para definir matemáticamente el cambio de temperatura, Bernoulli escribió textualmente: «El calor puede considerarse como un aumento en el movimiento de las partículas».
Explicó que la temperatura del gas es proporcional al cuadrado de la velocidad de sus partículas (
«Dado que es admitido que un mayor calor corresponde a un movimiento más intenso de las partículas, la elasticidad del aire se incrementa no solo por la condensación, sino también por el calor introducido… El calor es proporcional al cuadrado de la velocidad de los corpúsculos». [4]
A pesar de la brillantez de su deducción matemática, la propuesta de Bernoulli cayó en el olvido durante 120 años. Los motivos fueron varios:
- La física de entonces no entendía cómo los choques podían ser perfectamente elásticos. Si las partículas chocaban entre sí millones de veces, la lógica de la época decía que debían perder energía por fricción y el gas terminaría enfriándose y colapsando, algo que no ocurría.
- La teoría del calórico (el calor como fluido) explicaba muy bien los experimentos térmicos cotidianos y dominó la química durante todo el siglo XVIII.
- El propio Bernoulli continuó sus investigaciones en la hidrodinámica de fluidos macroscópicos líquidos y no volvió a desarrollar su modelo molecular. [1, 4]
No fue hasta la década de 1850, con el nacimiento de la termodinámica moderna de la mano de Rudolf Clausius y Joule, que se rescató el polvo del libro de Bernoulli para reconocer que se había adelantado un siglo entero a la ciencia. [7, 8]
[1] https://www.southampton.ac.uk
[7] https://www.britannica.com
[8] https://ia801404.us.archive.org
La presión según Clausius
Daniel Bernoulli (1738) (Daniel Bernoulli) había demostrado que los impactos moleculares generaban presión (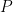

John Herapath (1821) redescubrió la teoría de forma independiente y fue el primero en intentar usar el momento lineal (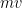

John James Waterston (1843) escribió un manuscrito asombroso en 1845 donde planteó, de manera prácticamente idéntica a la actual, la deducción de la presión y el factor 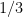
Rudolf Clausius es considerado el verdadero padre de la teoría cinética moderna. Tomando una idea simplificada que había publicado August Krönig en 1856, Clausius formalizó las matemáticas en 1857: definió correctamente los choques perfectamente elásticos en una caja, calculó el cambio de momento de la pared, e introdujo formalmente el promedio del cuadrado de las velocidades (
[2] https://centros.edu.xunta.gal
[7] https://www.studysmarter.es
[8] https://www.studysmarter.es
Este planteamiento fue más tarde consolidado y ampliado matemáticamente por James Clerk Maxwell y Ludwig Boltzmann entre 1859 y 1870 [1, 2, 3, 4]. La unificación de estos conceptos fue una respuesta a problemas fundamentales de la física del siglo XIX: [5]
- Superar la teoría del calórico: Hasta mediados del siglo XIX, se creía que el calor era un fluido invisible e indestructible llamado «calórico» que llenaba los espacios entre los átomos. Clausius propuso la teoría cinética del calor para demostrar que el calor no era una sustancia, sino la energía mecánica del movimiento (traslacional, rotacional y vibracional) de las propias moléculas.
- Explicar las Leyes de los Gases: Los científicos ya conocían de forma empírica la Ley de Charles y la de Gay-Lussac, que demostraban que la presión y el volumen aumentaban proporcionalmente con la temperatura (
). La única forma de justificar microscópicamente que un gas empujara con más fuerza las paredes al calentarse era si sus moléculas se movían más rápido (mayor energía cinética) a mayor temperatura.
- Consistencia con el Primer Principio de la Termodinámica: Los experimentos de James Prescott Joule demostraron que el trabajo mecánico podía convertirse en calor. Al conectar la temperatura directamente con la energía cinética molecular (
), la conservación de la energía cobraba pleno sentido: el calor introducido a un sistema se convertía de forma directa en el incremento de la energía cinética de sus partículas.
[3] https://repositorio-uapa.cuaed.unam.mx
[4] https://www.researchgate.net
[5] https://www.terpconnect.umd.edu
[6] https://www.visionlearning.com
[7] https://www.fceia.unr.edu.ar
[10] https://www.argumentos.us.es
[11] https://philsci-archive.pitt.edu
Fig. El físico matemático alemán Rudolf Clausius (1822-1888)
Sigamos el razonamiento de Clausius. Para deducir la ley de los gases ideales (
1. Hipótesis fundamentales
- El gas está formado por N moléculas puntuales de masa m.
- Las moléculas se mueven en direcciones aleatorias a velocidad v.
- Los choques con las paredes del contenedor de volumen V son perfectamente elásticos. [2, 3, 4, 5, 6]
2. El cambio de momento en un choque
Imaginemos un contenedor cúbico con paredes de área A y longitud L (donde 

- Su momento lineal inicial es:
- Tras el choque elástico, rebota con velocidad invertida:
- El cambio de momento (
) por choque es:


Por la tercera ley de Newton, la pared recibe un impulso de igual magnitud y signo opuesto:
3. Frecuencia de las colisiones
La molécula viaja una distancia de 
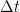
4. Fuerza y presión ejercida por una molécula
La fuerza (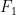
La presión (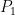
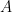
5. Presión total de todas las moléculas (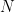
Para obtener la presión total (
Definimos el promedio del cuadrado de la velocidad en el eje X como 
Como el movimiento es tridimensional y totalmente aleatorio, no hay dirección preferencial:
Sustituimos 
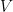
6. Conexión con la temperatura (Energía Cinética)
Multiplicamos y dividimos el segundo miembro por 2 para hacer explícita la energía cinética promedio (
En este punto ya es aparente que esta ecuación recuerda a la de los gases ideales, que era conocida en el siglo XIX. Para hacer completa la equivalencia entre ambas ecuaciones, Clausius introdujo el postulado central de la teoría cinética. Planteó que el movimiento de traslación de las moléculas se divide simétricamente en las tres direcciones del espacio tridimensional 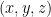
- Componente x:
- Componente y:
- Componente z:



Al sumar las tres componentes espaciales de la traslación, Clausius llegó a la famosa expresión de la energía cinética promedio de las moléculas como proporcional a la temperatura absoluta
donde 
Sustituimos 
7. Paso final a la forma macroscópica
Podemos expresar el número total de moléculas (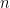
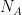

Dado que el producto de dos constantes (
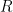

Cuando Clausius publicó su deducción en 1857, asumió por simplicidad matemática que todas las moléculas del gas se movían exactamente a la misma velocidad. James Clerk Maxwell (1859-1860) leyó el trabajo de Clausius y se dio cuenta de que era estadísticamente imposible que todas las moléculas viajaran a la misma velocidad, ya que los choques caóticos redistribuirían la energía constantemente. Maxwell tomó la ecuación de la presión de Clausius y le aplicó el cálculo de probabilidades, deduciendo la famosa curva de distribución de velocidades.
[3] https://galileo.phys.virginia.edu
[6] https://www.britannica.com
[8] https://es.khanacademy.org
[10] https://zientzia.eus
La Distribución de Maxwell-Boltzmann
Para deducir matemáticamente la distribución de velocidades de Maxwell-Boltzmann partiendo estrictamente de supuestos cinéticos y estadísticos, utilizaremos el elegante enfoque del propio James Clerk Maxwell (1860). Este método se basa en la simetría del espacio y en la independencia de los componentes de la velocidad en un gas ideal en equilibrio.

Fig. El físico teórico escocés James Clerk Maxwell (1831-1879)
1. Supuestos fundamentales
- El gas contiene un número inmenso de moléculas en movimiento caótico y estacionario (equilibrio térmico).
- Isotropía del espacio: No existe ninguna dirección espacial preferente. El gas se comporta igual en el eje
,
o
.
- Independencia estadística: La probabilidad de que una molécula tenga una velocidad determinada en el eje
2. La función de distribución en componentes
Definimos el vector velocidad de una molécula como 
Sea 



Dado que las velocidades en los tres ejes son independientes, la probabilidad conjunta de que una molécula tenga simultáneamente los valores 
3. Aplicando la isotropía espacial
Por otro lado, como el espacio es isótropo, esta probabilidad conjunta no puede depender de la dirección del vector, sino únicamente de su magnitud total 
Para resolver esta ecuación funcional, aplicamos el logaritmo natural en ambos lados:
Si derivamos parcialmente esta ecuación con respecto a 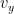

Dividiendo todo entre 
Si repetimos el mismo procedimiento derivando respecto a
Como el primer término depende exclusivamente de 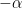
4. Resolviendo para una componente
Tomamos la ecuación para la componente
Integrando respecto a
donde 
5. Determinando las constantes (
La probabilidad total acumulada en todo el rango de velocidades posibles (de 
Utilizando la conocida integral gaussiana (

Para encontrar ahora el valor de la constante
El promedio del cuadrado de la velocidad
Resolviendo la integral por tablas estándar (
De la deducción de la ecuación de gases ideales según la teoría cinética, que vimos antes, sabemos que 
Sustituyendo
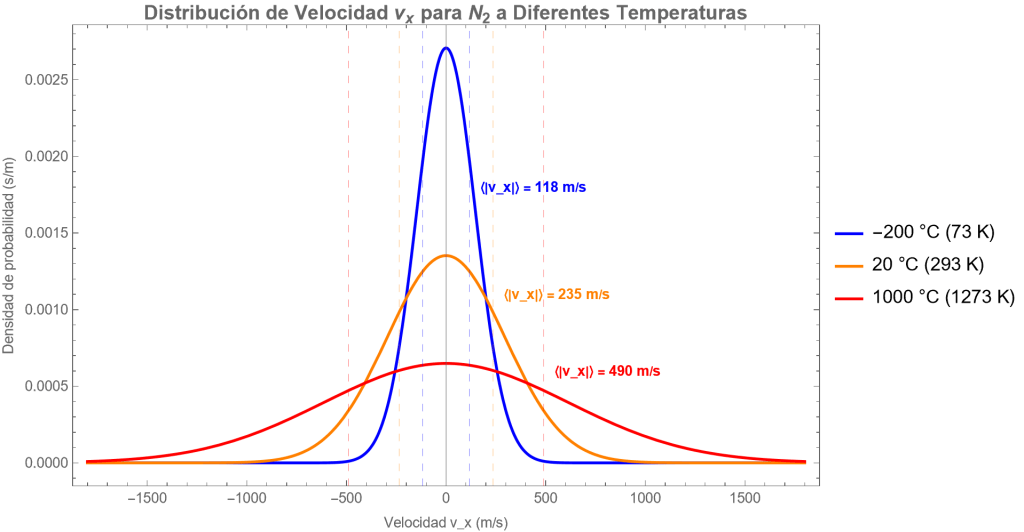
Fig. Distribución de vx para nitrógeno a -200, 20 y 1000 ºC. Se muestran también los valores respectivos de la rapidez media de las moléculas
6. Distribución de la rapidez tridimensional
Para conocer la probabilidad de la rapidez total
Para pasar de coordenadas cartesianas 

Sustituyendo este cascarón y simplificando, obtenemos finalmente la famosa Distribución de Rapideces de Maxwell-Boltzmann: [1]
A partir de esta bonita función matemática, es posible calcular tres velocidades críticas para un gas: la velocidad más probable, la velocidad promedio y la velocidad cuadrática media (
[2] https://es.khanacademy.org
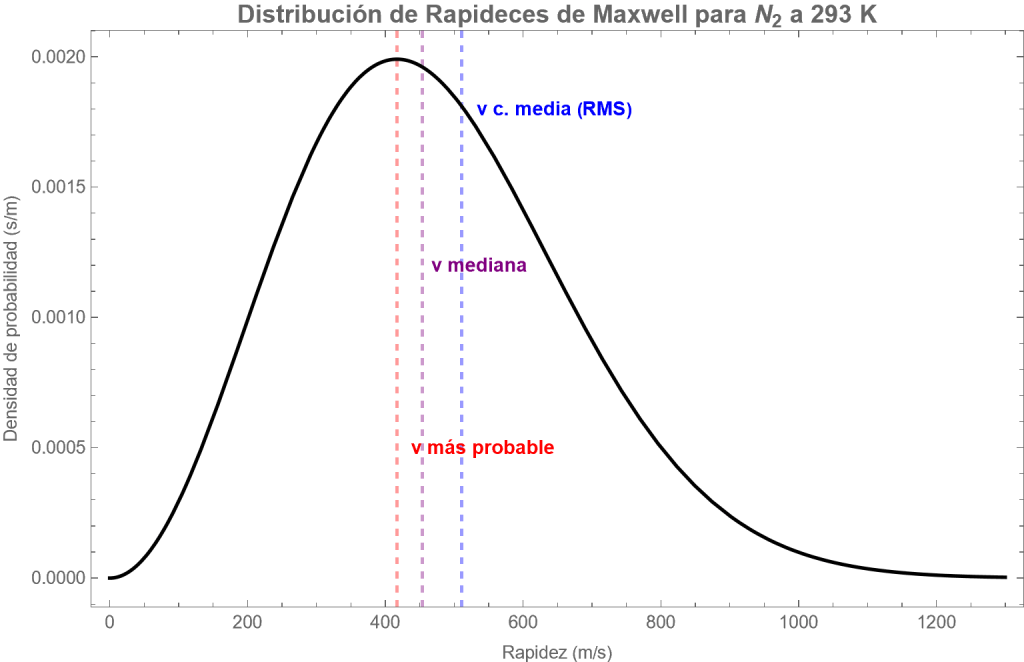
Velocidad más probable de las moléculas de un gas
Para calcular la velocidad más probable (
Para facilitar el álgebra, agrupamos todas las constantes externas en una sola letra 

Derivamos 
![\frac{d}{dv}[u \cdot v] = u'v + uv'](https://s0.wp.com/latex.php?latex=%5Cfrac%7Bd%7D%7Bdv%7D%5Bu+%5Ccdot+v%5D+%3D+u%27v+%2B+uv%27&bg=ffffff&fg=000&s=0&c=20201002)
- Sea
- Sea


Combinando los términos:
Factorizamos el término común 
Ahora tenemos en cuenta que en los máximos (y en los mínimos) de una función, la pendiente es nula, esto es, la derivada es cero:
Para que la expresión anterior sea cero, alguno de los dos factores debe ser cero. Existen tres soluciones matemáticas posibles:
(Este es un mínimo físico: la probabilidad de que una molécula esté completamente estática es cero).
(Otro mínimo: la probabilidad de tener velocidad infinita es cero).
(Este término representa el punto máximo de la curva).
Tomamos la tercera condición que define el máximo: 



Recordamos que definimos originalmente
Si multiplicamos el numerador y el denominador por el número de Avogadro (
Como ejemplo práctico, con esta fórmula se puede calcular que una molécula de nitrógeno (

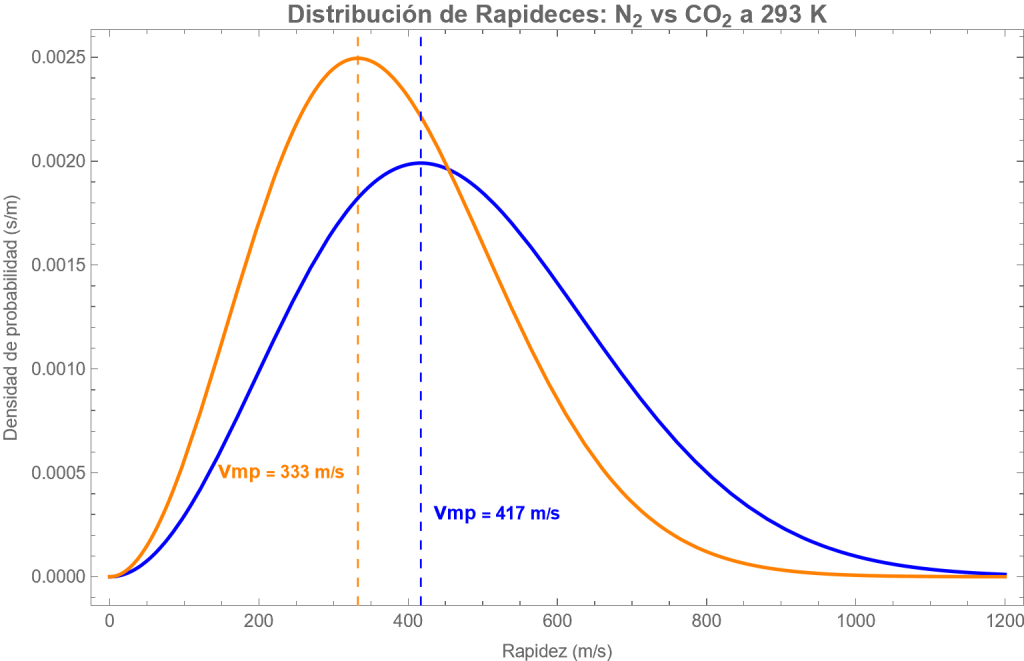
Fig. Distribución de rapideces moleculares del CO2 (rojo) y N2 (azul) a 20ºC. Se indican también sus respectivas velocidades más probables. El CO2 es más pesado que el N2
La velocidad media y la velocidad cuadrática media de las moléculas de un gas
Para calcular la velocidad promedio (o rapidez media, denotada como 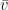

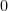

Sustituimos la función de distribución de Maxwell-Boltzmann en la ecuación del promedio:
Para simplificar el álgebra, extraemos las constantes fuera de la integral y definimos de nuevo la constante del exponente como
Nos enfocamos ahora en resolver la integral definida:
Hacemos un cambio de variable estándar para eliminar el exponente cuadrático:
- Sea
- Reescribimos el término
como
- Los límites de integración se mantienen igual (de


Sustituyendo estos términos, la integral se transforma en:
Esta es una integral estándar de la función Gamma (o se puede resolver fácilmente por partes), cuyo resultado analítico es de la forma 
Ahora que conocemos el valor de la integral (
Desarrollamos el exponente fraccionario 
Simplificamos las variables equivalentes (el 

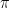
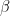
Finalmente, deshacemos el cambio de la constante sustituyendo
Si multiplicamos tanto el numerador como el deminador por el número de Avogadro (
Si comparamos numéricamente los coeficientes bajo la raíz en la velocidad más probable (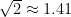

Un cálculo similar permite obtener la velocidad cuadrática media de las moléculas de un gas (conocida como 
Al sustituir la distribución de Maxwell en esta ecuación, se obtiene una integral de tablas gaussianas del tipo:
cuya solución final es:
Como el objetivo inicial era encontrar
Expresado en variables macroscópicas utilizando la constante universal de los gases (
Resumen y comparación de las tres velocidades
Ahora que hemos calculado las tres velocidades de la distribución de Maxwell-Boltzmann, podemos ordenarlas según sus coeficientes numéricos bajo la raíz:
- Velocidad más probable (
):
(El pico más alto de la curva).
- Velocidad promedio ($\bar{v}$):
(El promedio aritmético desviado por la «cola» derecha).
- Velocidad cuadrática media (

Esta última es la velocidad estadística asociada directamente a la energía cinética y a la presión.
Algunas consecuencias de la distribución de Maxwell
1. Por qué la ropa se seca y los charcos desaparecen?
Si el agua necesita llegar a 100 °C para hervir y evaporarse, ¿por qué el agua de un charco en la calle o la ropa tendida se secan a solo 20 °C?
Explicación: A 20 °C, la velocidad promedio de las moléculas de agua no basta para romper los enlaces líquidos. Sin embargo, gracias a la curva de Maxwell-Boltzmann, siempre hay una pequeña fracción en la «cola derecha» (las moléculas ultra-rápidas) que tienen energía suficiente para escapar al aire. Al irse las más rápidas, el promedio del charco baja (el charco se enfría ligeramente), pero el sol vuelve a calentar el charco, redistribuyendo las velocidades y creando una nueva «cola rápida». El proceso se repite hasta que el charco desaparece.
2. Por qué el gas de la risa (el helio) te cambia la voz?
Es sabido que aspirar el helio de un globo te da voz de ardilla, pero por qué?
Explicación: La voz humana depende de cómo resuena el aire en las cuerdas vocales. Como el Helio es 7 veces más ligero que el Nitrógeno del aire, la fórmula de la velocidad de Maxwell (
Ahora bien, en gases con moléculas monoatómicas como los gases nobles (Helio, Neón, Argón,…) la onda de presión del sonido se mueve al 75% de la velocidad de sus moléculas (en gases diatómicos como O2, N2, H2, etc., sería al 68%). Esto implica que la velocidad del sonido en el gas helio es de unos 1000 m/s, casi tres veces mayor que en aire.
Supongamos que un tenor está cantando un Do4 (C4 o «Do de pecho»), a una temperatura ambiente estándar de 20 °C 
- Frecuencia del Do4 (
):
- Velocidad del sonido en el aire
):


La longitud de onda 

Como 
Una frecuencia de 

3. La energía de activación
¿Por qué un bosque no se incendia solo a pesar de estar rodeado de oxígeno, o por qué una tarta no se hornea sola en la encimera?
Explicación: Para que ocurra una reacción química (como la combustión o el cocinado), las moléculas deben chocar con una energía mínima llamada Energía de Activación. Al graficar este umbral sobre la curva de Maxwell, se entiende visualmente que a temperatura ambiente casi ninguna molécula da la talla. Al encender un fósforo o encender el horno, la curva se desplaza y se achata hacia la derecha, haciendo que de golpe millones de moléculas crucen la línea de meta y comiencen a reaccionar.
La figura siguiente muestra, en escala logarítmica, la probabilidad de que una molécula tenga una cierta energía, para ambas temperaturas, 20 y 200 grados, así como la energía típica de activación de la reacción de Maillard en el pastel (línea vertical). Se trata de la reacción entre un azúcar reductor y un aminoácido (proteína) de la leche, huevo o harina, clave del horneado.
Si calculamos el área de ambas curvas para energías superiores a la de oxidación podemos obtener la fracción de moléculas que están por encima de la energía de oxidación: A 20 °C (Temperatura ambiente) la fracción es de apenas 

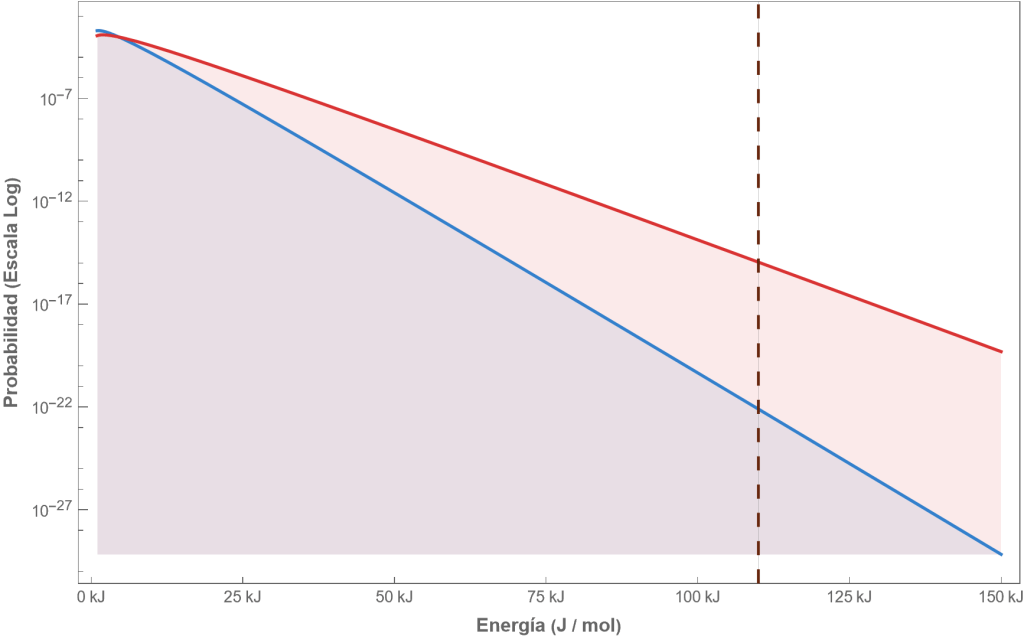
Fig. Distribución de energías de las moléculas aire dentro de un horno a 20 y 200 grados Centígrados, y energía de activación de las reacciones de caramelización
Al aumentar la temperatura la cola de la distribución se desplaza y aumenta exponencialmente el número de moléculas de energía superior a la de activación. Esto es lo que causa que la velocidad de reacción venga dada por la expresión de Arrhenius:
donde k (Constante de velocidad): Indica qué tan rápido ocurre la reacción química, A (Factor preexponencial o factor de frecuencia): Representa la frecuencia total de colisiones entre las moléculas y la probabilidad de que choquen con la orientación geométrica correcta, 


En un artículo futuro estudiaremos cómo se obtiene la ecuación de Arrhenius a partir de la Teoría Cinética.
Debido a la naturaleza exponencial de la ley de Arrhenius, un incremento de temperatura no duplica la velocidad; en la reacción de Maillard, la tasa de reacción se duplica aproximadamente por cada 10 °C de aumento. Pasar de 20 °C a 160 o 200 °C acelera el proceso miles de veces, permitiendo que ocurra ante nuestros ojos en lugar de en tiempos astronómicos.

Deja un comentario