
Estudiaremos aquí algunos elementos que aportó Ludwig Boltzmann a la Mecánica Estadística y que generalizaron las aportaciones de James Clerk Maxwell. Para ampliar el trabajo de Maxwell, que solo estudiaba la velocidad de las partículas libres de un gas (La Teoría Cinética y la Distribución de Maxwell), hasta descubrir el factor de Boltzmann (
Su aportación se basó en dos pilares: primero, introdujo las fuerzas externas (como la gravedad) que pueden actuar sobre un gas; segundo, y lo más revolucionario, unió la probabilidad matemática con las colisiones mecánicas que se producen entre las moléculas.
El decrecimiento hidrostático de la densidad con la altura
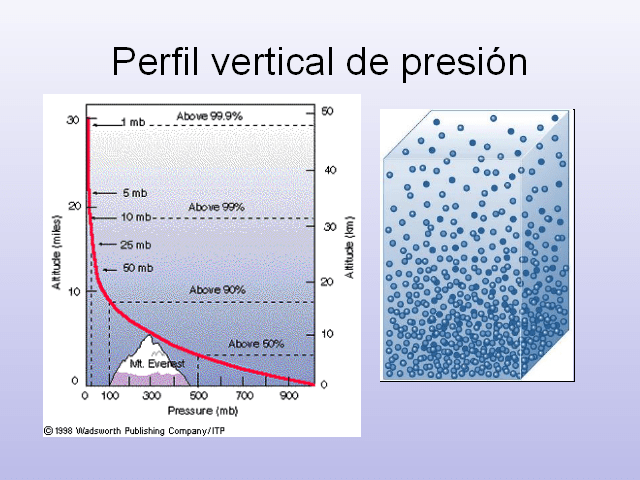
Sabemos que la presión y la densidad de la atmósfera disminuyen con la altura 

Históricamente se considera que Edmond Halley fue el primero en deducir en 1686 una fórmula barométrica que relaciona la presión y también la densidad exponencialmente con la altura. [1]
Para realizar la deducción, Halley no utilizó el cálculo integral moderno (que acababa de ser inventado de forma independiente por Newton y Leibniz y aún no era de uso común), sino que utilizó la geometría analítica y las propiedades de las curvas logarítmicas. Unió dos piezas fundamentales del rompecabezas:
- La Ley de Boyle-Mariotte (1662): Sabía que el volumen de una masa de aire es inversamente proporcional a la presión, lo que significa que la densidad del aire cambia proporcionalmente con la presión.
- El principio de Torricelli y Pascal (1643-1648): Sabía que la atmósfera tiene peso y que la presión a una determinada altura equivale al peso de la columna de aire que queda por encima. [1, 3, 4, 5, 6]
Combinando ambos supuestos de forma geométrica, Halley demostró que si la altitud aumenta en progresión aritmética (


Fue el gran matemático francés Pierre-Simon de Laplace quien, en 1805, tomó la deducción de Halley y la tradujo al lenguaje del cálculo diferencial moderno, introduciendo las variables físicas macroscópicas que usamos hoy en día: [7]
donde 
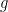
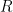
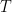
[4] https://www.britannica.com
[5] https://pubmed.ncbi.nlm.nih.gov
Para deducir la fórmula barométrica (
1. El equilibrio hidrostático (Mecánica de fluidos)
Aislamos una rebanada horizontal de aire a una altura 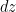
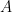

- Fuerza hacia abajo debida a la presión del aire que tiene encima:
- Fuerza hacia arriba debida a la presión del aire que tiene debajo:
- Fuerza hacia abajo debida a su propio peso:
(donde $latex \rho$ es la densidad de masa del gas).


Para que la rebanada no se mueva, la suma de fuerzas debe ser cero (
Cancelamos el área
Esta es la ecuación fundamental de la hidrostática. Nos dice que la presión disminuye al aumentar la altura porque hay menos peso de aire encima.
2. Conectar la densidad de masa (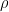
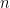
La densidad de masa (
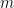

Sustituyendo esto en la ecuación hidrostática:
3. Aplicar la ley de los gases ideales
Sabemos que la ley de los gases ideales a nivel molecular se escribe como (La Teoría Cinética y la Distribución de Maxwell):
Suponiendo que la atmósfera fuera isoterma (la temperatura
4. Resolver la ecuación diferencial por separación de variables
Ahora igualamos las dos expresiones que tenemos para 
Separamos las variables dejando todos los términos con
Integremos ambos lados de la ecuación. Definimos que a la altura del suelo (

Resolvemos las integrales básicas (
Para despejar 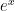
Llegamos finalmente a la ecuación deducida:
Como la presión es directamente proporcional a la densidad (
Cuando T varía con la altura, como es el caso de la atmósfera de la Tierra, la presión responde a una función de la forma:
Donde 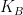

En la troposfera (desde el suelo hasta unos 



La integración matemática en esta capa no nos da entonces una exponencial pura, sino una función potencial debido al gradiente lineal:
En la estratosfera (desde los 
Al aumentar
[2] https://www.studysmarter.es
[5] https://edea.juntadeandalucia.es
[6] https://www.studysmarter.es
[7] https://www.studysmarter.es
probabilidad de La Altura de una Molécula en la Atmósfera
Maxwell había deducido que la probabilidad de una velocidad era 
1. El primer paso: Identificar la energía potencial (1868)
En la fórmula hidrostática del decrecimiento de la densidad con la altura 
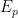

Combinando la distribución de velocidades de Maxwell (energía cinética, 

Para escribir esta función de distribución, llamada de Maxwell-Boltzmann, de forma normalizada, la integral de la función sobre todo el espacio de fases (todas las posiciones y velocidades posibles) debe ser igual a 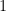
Asumiendo un volumen cerrado de altura infinita sometido a un campo gravitatorio uniforme $V(z) = mgz$, la función de densidad de probabilidad conjunta para la posición 

Donde
A continuación se detalla cómo se obtiene la constante de normalización para el espacio completo. La suma de todas las probabilidades debe ser la unidad:
La función original tiene la forma 
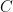
Cada una de las tres integrales de velocidad de la componente cartesiana se resuelve mediante la identidad de la integral de Gauss 
Al estar en tres dimensiones (
La integración sobre las coordenadas horizontales 
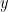

La integración sobre la altura 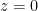
Multiplicamos todos los componentes calculados:
Despejando
Sustituyendo la constante
Esta función permite calcular la probabilidad exacta de encontrar una molécula de masa
Cuando Boltzmann escribe esta ecuación para la probabilidad de una molécula, se refiere a la densidad de probabilidad de encontrar la molécula por unidad de volumen del espacio de fases. La probabilidad de encontrar a la molécula en el diferencial de volumen 
Donde 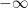
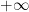
Separamos la ecuación en su parte espacial y su parte cinemática:
Esa triple integral es una integral gaussiana. Su resultado es un valor puramente numérico que depende de la temperatura, pero que es idéntico para cualquier altura
Al resolver las tres direcciones de la velocidad, la triple integral da como resultado:
Ahora sustituimos ese resultado de vuelta en nuestra ecuación. Todo ese bloque matemático proveniente de las velocidades se absorbe dentro de una nueva constante de normalización (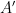
Si asumimos que la atmósfera es homogénea en el plano horizontal (las coordenadas 

que nos dice que la probabilidad de que una molécula esté a una cierta altura depende de su energía potencial de la misma forma que la presión hidrostática y que la densidad.
2. La generalización: El equilibrio en las colisiones (1872)
El paso definitivo de Boltzmann fue demostrar que esto no solo aplicaba a la gravedad, sino a cualquier sistema de partículas enlazadas o interactivas (gases poliatómicos, líquidos o sólidos). Para ello, analizó qué ocurre matemáticamente debido a los choques.
Imaginemos un sistema que puede saltar entre un «Estado 1» (con energía 
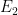
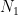

Del mismo modo, la tasa de regreso es:
Para que el sistema alcance el equilibrio térmico, ambas tasas deben igualarse exactamente (un principio conocido como equilibrio detallado):
3. La conexión de la entropía termodinámica S con el número de microestados 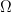
La termodinámica clásica ya había definido, desde Clausius, la variación de entropía (
Para un gas ideal en una expansión isotérmica (temperatura 
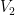
Donde 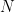
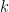
Boltzmann postuló que un macroestado del gas (definido por variables como presión, volumen y temperatura) está compuesto por una inmensa cantidad de microestados compatibles (posiciones y velocidades específicas de cada molécula). [2]
Si consideramos una sola partícula que pasa de un volumen 

Si aplicamos el logaritmo natural a ambos lados de la ecuación obtenemos:
Al despejar el término del volumen e igualarlo con la ecuación del macroestado de Clausius, Boltzmann descubrió una proporcionalidad directa entre la entropía y el número de microestados:
Esto se traduce formalmente como:
Boltzmann determinó que la función matemática idónea debía ser el logaritmo debido a las propiedades esperables del sistema:
- La entropía macroscópica es una propiedad aditiva (
).
- La probabilidad o número de microestados combinados es multiplicativa (
). [3, 4]
La única función matemática que transforma una multiplicación en una suma es el logaritmo:
Esto consolidó de manera abstracta la constante de proporcionalidad k, dando lugar a la célebre ecuación que conecta las propiedades microscópicas del gas con las leyes macroscópicas de la energía:

Foto. Tumba de Ludwig Boltzmann en Viena, donde aparece grabada su famosa relación entre la entropía y el número de microestados
4. La conexión con las leyes mecánicas
Las leyes de la mecánica clásica (como la simetría de las colisiones elásticas bajo reversión temporal) dictan que la probabilidad de que ocurra una colisión que intercambie energía depende fundamentalmente de la energía disponible en las demás partículas del sistema, que actúan como un baño térmico. Boltzmann demostró que la relación entre estas tasas de transición mecánica tiene esta forma:
Esto se puede demostrar del modo siguiente:
Imaginemos que nuestro sistema de partículas está en contacto con un gran baño térmico a temperatura 


La probabilidad de que el sistema realice una transición depende de cuántos microestados (configuraciones) tiene disponibles el baño térmico para acomodar ese cambio de energía. Si llamamos 
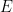
Boltzmann había demostrado (sección anterior) que la entropía termodinámica de un sistema de energía fija E tiene que estar relacionada con el número de microestados compatibles con esa energía, mediante la expresión 

Como el baño térmico es gigantesco en comparación con nuestro pequeño sistema de una sola molécula (o unas pocas moléculas), las energías 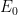
Si restamos las dos entropías que aparecen dentro del exponente de nuestra expresión, el término base 
Por definición fundamental de la termodinámica clásica y estadística, el cambio de la entropía con respecto a la energía interna es el inverso de la temperatura absoluta:
Sustituyendo esta identidad en la ecuación anterior, queda:
Introducimos este resultado directamente en el exponente de nuestra ecuación:
Reorganizando el signo menos en el numerador para que coincida exactamente con la estructura de nuestra fórmula original:
Sustituyendo esto en la relación de poblaciones (
Para que esta igualdad se cumpla para cualquier par de estados arbitrarios en el universo, la población (o probabilidad 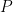
El análisis estadístico de los macroestados
Años más tarde, Boltzmann completó su razonamiento analizando no sólo microestados de moléculas individuales y su probabilidad, sino macroestados, que son estados de un sistema macroscópico con alguna propiedad macroscópica definida (por ejemplo, energía conocida). Demostró que si un estado macroscópico tiene una energía 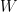
Al integrar el peso estadístico en el análisis, la probabilidad real de que midamos un sistema en un estado de energía específico es el producto de «cuántas opciones tiene» (peso estadístico,
Vimos en la primera sección que la probabilidad de encontrar a una molécula atmosférica con una energía E era 
Si integramos ahora para todas las velocidades, obtenemos la probabilidad de que la molécula tenga cualquier velocidad pero una altura determinada z: 
Pero si tenemos un estado macroscópico (macroestado) que sabemos que tiene energía E y que es compatible con muchos microestados no observables, e integramos manteniendo E constante: Agrupamos todos los microestados que sumen esa energía, y ahí es donde aparece explícitamente el peso estadístico: 
Esta fórmula es de las más usadas en la física estadística moderna: rige desde la electrónica de los semiconductores en un teléfono móvil hasta las reacciones químicas en nuestras células.
Un procedimiento general para operar con macroestados fue sistematizado por Josiah Willard Gibbs en su obra cumbre, «Elementary Principles in Statistical Mechanics» («Principios elementales de la mecánica estadística»), publicada en 1902, donde introdujo los formalismos de los conjuntos microcanónico, canónico y macrocanónico.
La Tendencia Física hacia los Estados más Probables
La relación 
Ludwig Boltzmann no tenía ninguna evidencia matemática ni experimental sólida para demostrar la hipótesis ergódica. [1, 2, 3] Para Boltzmann era una necesidad lógica y un postulado de trabajo indispensable para que sus ecuaciones de probabilidad tuvieran sentido físico. [2, 4] Su teoría predecía perfectamente las propiedades medibles de los gases (como la presión, el calor específico y la viscosidad), lo que significaba que su audaz suposición sobre el espacio de fases tenía que ser correcta en la práctica.
Antes de proponer formalmente la hipótesis ergódica, Boltzmann intentó demostrar el aumento de la entropía mediante mecánica pura con su famoso Teorema H (1872). Definió una función matemática matemática 
Varios físicos demostraron que la mecánica clásica de Newton chocaba directamente con la idea de que un sistema visita «ciegamente» todos los estados aumentando siempre la entropía:
Crítica de Loschmidt (1876): Las leyes de Newton son reversibles en el tiempo. Si filmas las colisiones de las partículas de un gas y reproduces el video al revés, las ecuaciones siguen siendo válidas. Si la mecánica permite que la entropía disminuya al ir hacia atrás, ¿por qué en el mundo real solo la vemos aumentar? [10]
Crítica de Poincaré (1890) y Zermelo (1896): El teorema de recurrencia de Poincaré demostraba que cualquier sistema mecánico cerrado de partículas, tarde o temprano, volverá a una configuración tan cercana como se quiera a su estado inicial. Por lo tanto, la entropía no puede crecer indefinidamente; tendría que fluctuar, subiendo y bajando.
Ante estas críticas, Boltzmann tuvo que admitir que el aumento de la entropía y la exploración del espacio de fases no eran leyes mecánicas absolutas, sino leyes estadísticas. [2, 11] Fue en este punto donde acuñó formalmente el concepto de sistema «ergódico» (alrededor de 1884). Admitió [2, 12] que las caóticas y complejas trayectorias del sistema terminarían pasando cerca de cada punto de la superficie de energía constante del espacio de fases. [2]. Pero calculó el tiempo que tardaría un gas en regresar cerca de su estado inicial, y demostró que para un simple centímetro cúbico de gas, el tiempo de retorno es muchísimo mayor que la edad del universo. Es famoso su comentario en un debate: «Ustedes esperen [a que el sistema regrese a su estado inicial], yo no viviré tanto».
Décadas después de su muerte, los matemáticos Ernst Plancherel y Artur Rozenthal (1913) demostraron que la hipótesis ergódica estricta de Boltzmann era matemáticamente imposible para sistemas físicos reales: una línea continua unidimensional no puede llenar un espacio tridimensional o de más dimensiones sin cruzarse consigo misma. Y este cruce es imposible en un sistema mecánico, pues implicaría que un mismo estado inicial genera dos evoluciones temporales diferentes, en contra de las leyes y las evidencias de la mecánica clásica [2, 3].
Sin embargo, matemáticos como John von Neumann y George Birkhoff salvaron la física estadística en la década de 1930 al demostrar la hipótesis cuasi-ergódica: el sistema no pasa exactamente por todos los estados compatibles con la energía inicial, pero pasa tan cerca como se quiera de casi todos ellos, permaneciendo en cada región del espacio de fases un tiempo estrictamente proporcional al volumen de esa región [13, 14] (Von Neumann, G. D. Birkhoff, Proof of the Ergodic Theorem with Proof of the Quasi-Ergodic Hypothesis with Physical, 1931-1932).
Dado que el sistema pasa «tan cerca como se quiera» de casi cualquier punto del espacio de fases, la configuración donde todas las moléculas de gas se agrupan en una esquina de la caja contenedora está incluida en la evolución del sistema. Es un estado macroscópico perfectamente válido. Pero el teorema de von Neumann-Birkhoff establece que el tiempo que el sistema pasa en una región es estrictamente proporcional al volumen de esa región en el espacio de fases. El «volumen» (el número de microestados
Por lo tanto, si tuvieramos que esperar a que ocurra de forma espontánea en un gas macroscópico (por ejemplo, un simple globo de aire), el tiempo medio de espera superaría con creces la edad actual del universo. Y si por un azar infinitesimal el sistema cayera en esa región de «todas las partículas en la esquina», saldría de ella de forma casi inmediata. Las partículas chocan y se repelen, moviéndose a cientos de metros por segundo hacia el espacio vacío. El sistema «escaparía» de ese microestado ultra-ordenado en una fracción de picosegundo, destruyendo la fluctuación antes de que cualquier aparato macroscópico pudiera registrarla.
El estado que llamamos «equilibrio térmico» no es un estado estático; es un océano de microestados homogéneos. Alrededor de cualquier fluctuación grande pero brevísima, el sistema pasará eones fluctuando de formas imperceptibles (pequeñas variaciones de densidad o temperatura en regiones microscópicas). Para un observador humano o un termómetro ordinario, el sistema parecerá perfectamente estable y en calma chicha antes y después de esas fluctuaciones aberrantes. [1]
Este panorama demuestra que el tiempo y el espacio de fases están conectados de forma simétrica: lo que ocupa poco espacio en la geometría del universo microscópico, ocupa también una milésima de pestañeo en la historia del tiempo. Boltzmann intuyó esto de forma brillante, y los teoremas de los años 30 simplemente le pusieron el sello de rigor matemático que sus oponentes le exigían.
Boltzmann tenía razón en su intuición física y estadística, aunque no llegó a conciliar rigurosamente su intuición con las leyes de la mecánica clásica.
[1] https://www.sciencedirect.com
[3] https://plato.stanford.edu
[4] https://ui.adsabs.harvard.edu
[5] https://www.thphys.uni-heidelberg.de
[9] https://news.web.baylor.edu
[12] https://www.instagram.com
[14] https://dokumen.pub

Deja un comentario