
La fórmula de Arrhenius establece matemáticamente cómo cambia la constante de velocidad de una reacción química en función de la temperatura y se expresa como: [1]
Donde 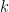
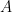
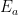
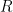

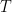

Esta ecuación es fundamental porque conecta la termodinámica con la cinética química, permitiendo predecir exactamente cómo la temperatura y la energía molecular influyen en la velocidad a la que ocurren las transformaciones químicas. La ecuación de Arrhenius transformó la química teórica y práctica por varias razones:
- Explica cuantitativamente por qué un pequeño aumento de temperatura acelera drásticamente una reacción (al aumentar el número de moléculas que superan la barrera
- Permite a los científicos aislar y calcular la energía de activación
- Demuestra de manera matemática el funcionamiento de los catalizadores, los cuales aceleran las reacciones disminuyendo el valor de
- Es crucial para diseñar reactores químicos seguros, estimar la fecha de caducidad de alimentos o fármacos, y modelar procesos geoquímicos o atmosféricos.
[1] https://www.studysmarter.es
[2] https://proyectodescartes.org
[3] https://success.openhealth.fr
[5] https://theory.labster.com
[7] https://www.studysmarter.es
En varios artículos previos explicamos cómo surgieron algunos de los conceptos más importantes de la teoría cinética de los gases y de la materia en general: La Teoría Cinética y la Distribución de Maxwell; De la distribución de Maxwell a la probabilidad de Boltzmann; Entropía, irreversibilidad y flecha del tiempo según Gibbs, y la importancia que tuvo la teoría cinética para consolidar la teoría atómica de la materia (Por qué los Científicos Empezaron a Creer en los Átomos: El Periodo 1808 – 1908). En este artículo mostraremos cómo la ecuación de Arrhenius de la química se puede deducir también de la teoría cinética.
Para obtener la ecuación de Arrhenius a partir de la teoría cinética, debemos dividir la deducción en dos grandes bloques matemáticos: Parte 1, donde calculamos la frecuencia de colisión total (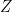
Parte 1: Deducción de la Frecuencia de Colisión Z
Imaginemos un gas compuesto por dos tipos de moléculas esféricas, 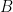




Paso 1.1: Definir la Sección Eficaz
Dos moléculas chocarán si la distancia entre sus centros es menor o igual a la suma de sus radios (
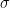
Paso 1.2: El Cilindro de Colisión
Consideremos una sola molécula 
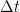
Cualquier molécula
Paso 1.3: Colisiones de una sola molécula A con todas las B
El número de moléculas
Para obtener la tasa de colisión por unidad de tiempo (
Paso 1.4: Frecuencia de colisión total por unidad de volumen (ZAB)
Como en nuestro volumen real no hay una sola molécula
Paso 1.5: Obtener la velocidad relativa media entre dos clases de moléculas
Para demostrar rigurosamente la expresión de la velocidad relativa media (
(i) Definir las funciones de distribución individuales
Consideremos dos gases ideales, 



Según la estadística de Maxwell-Boltzmann, la probabilidad de encontrar una molécula de la especie
Del mismo modo, para la especie
Dado que los movimientos de
(ii) El cambio de variables (Centro de Masas y Velocidad Relativa)
Para hallar la velocidad relativa, definimos el vector de velocidad relativa ($\latex vec{v}_{rel}$) y el vector de velocidad del centro de masas (
Ahora debemos reescribir la energía cinética total del exponente, 
Donde hemos introducido dos nuevos parámetros físicos, la masa total (

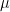

(iii) Transformar el diferencial (El Jacobiano)
Al cambiar las variables de integración de 

Sustituyendo la energía del Paso 2 y el diferencial en nuestra probabilidad conjunta, la función de distribución se factoriza de forma limpia en dos partes independientes:
Este paso es el más bello matemáticamente: nos demuestra que el movimiento del centro de masas y el movimiento relativo se comportan como si fueran dos partículas ficticias independientes. La partícula del movimiento relativo tiene una masa igual a la masa reducida (
(iv) Calcular el valor medio de la velocidad relativa
La velocidad relativa media 

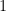
Pasamos a coordenadas esféricas en el espacio de velocidades relativas, donde el diferencial de volumen es 



(v) Resolver la integral estándar
Para resolver la integral que aparece en esta expresión, hacemos un cambio de variable estándar: 

Sustituimos este resultado en la ecuación del Paso 4:
Agrupamos los términos bajo una sola raíz cuadrada y simplificamos los coeficientes:
💡 Corolario físico interesante:
Si las dos moléculas que chocan son del mismo gas, entonces 


Esto demuestra que, para un mismo gas, la velocidad relativa media entre sus moléculas es exactamente 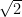
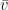
Sustituyendo 
Parte 2: El Filtro Energético y la Ecuación de Arrhenius
No todos los choques de 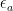

Paso 2.1: Aplicar la Distribución de Energías
Según la mecánica estadística, la probabilidad de que una colisión ocurra con una energía cinética relativa 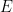
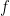
Paso 2.2: Resolver la integral
Esta integral es directa. La antiderivada de 

Como 
Multiplicando el numerador y el denominador del exponente por el número de Avogadro (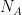


Paso 2.3: Unir Frecuencia, Orientación y Energía
La velocidad de la reacción (
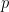
En la cinética química macroscópica, la ley de velocidad para una reacción 

Paso 2.4: Identificación con la expresión de Arrhenius
El término entre paréntesis agrupa todos los parámetros físicos y la dependencia débil de la temperatura (
Esta ecuación fue obtenida de forma experimental (empírica) por Svante Arrhenius en 1889, aunque se basó en deducciones teóricas previas del químico Jacobus Henricus van ‘t Hoff sobre la dependencia de la velocidad de reacción con la temperatura [1]. Fueron Max Trautz y William Lewis (1916) quienes utilizando la teoría cinética de los gases, demostraron matemáticamente que el factor preexponencial (A) se corresponde con la frecuencia de los choques moleculares y su orientación geométrica, tal como lo hemos demostrado aquí.

Foto. El físico y químico sueco Svante August Arrhenius (1859-1927), premio Nobel de química en 1903.
El factor estérico p lo hemos incluido en la constante A, sin explicitarlo teóricamente, porque deducirlo matemáticamente es sumamente difícil debido a que las moléculas reales no son esferas perfectas. Para obtenerlo, se suele operar empíricamente: [1, 2]
- Se mide experimentalmente la constante de reacción k (última expresión del paso 2.3) a distintas temperaturas y se grafica, generalmente en forma logarítmica. Esto permite deducir la constante
- Se calcula teóricamente la sección eficaz de colisión molecular
- Se halla el factor estérico real despejándolo como el cociente de la constante obtenida en el paso 1 y la

Este factor estérico se observa que varía según el tipo de molécula reaccionante:
Átomos o moléculas esféricas simples (
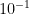

Moléculas de complejidad moderada (
Moléculas complejas o biomoléculas (
Reacciones de arpón (
Moléculas con grados de libertad internos
Las moléculas poliatómicas tienen en general grados de libertad internos, como modos de vibración y rotación. En estos casos, la energía necesaria para romper un enlace no proviene únicamente de la velocidad con la que chocan de frente (energía cinética traslacional). La molécula puede acumular y redistribuir energía en sus múltiples grados de libertad internos, que incluyen la rotación y, sobre todo, las vibraciones de sus múltiples enlaces.
El químico Cyril Hinshelwood (Premio Nobel de Química) resolvió este problema modificando la teoría de colisiones. Si la energía puede repartirse en múltiples «términos cuadráticos» de energía (cinética y potencial de vibración), la integral de Maxwell-Boltzmann cambia por completo y la probabilidad de éxito de la colisión aumenta drásticamente.

Foto. El químico inglés Cyril Norman Hinshelwood (1897-1967), premio Nobel de química en 1956.
A continuación se detalla la demostración de cómo se modifica matemáticamente la integral.
1. Planteamiento del problema con s grados de libertad
En una colisión simple de esferas rígidas, solo importa la energía cinética a la largo del eje de impacto. Esto equivale a 2 términos cuadráticos de energía (la velocidad de cada molécula en ese eje, 
Si una molécula poliatómica grande tiene 
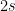
2. Obtención de la nueva función de distribución de probabilidad
Según la mecánica estadística clásica (teorema de equipartición), la probabilidad de que un sistema con 
Para deducir la función de distribución de probabilidad 

El principio de equirrepartición nos dice que cada término cuadrático de energía (ya sea de momento 


A continuación, deduciremos paso a paso cómo la geometría de ese espacio multidimensional de energías da origen a la distribución Gamma.
Paso 1: El espacio de fases y los términos cuadráticos
Imaginemos que la energía total 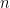
Aquí, cada 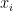

Paso 2: Calcular el volumen del espacio accesible (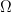
Según la hipótesis fundamental de la mecánica estadística de Boltzmann, el número de microestados disponibles (el volumen en el espacio de fases
El volumen de una hiperesfera de radio 
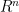
Como tenemos
Paso 3: Densidad de estados (El factor geométrico 
La densidad de estados 
Ignorando las constantes multiplicativas (que luego absorberá la constante de normalización), el factor de degeneración geométrica de los estados es:
Este resultado es clave: nos dice que a medida que la energía aumenta, el espacio geométrico disponible para acomodar esa energía crece de forma polinómica según el número de grados de libertad.
Paso 4: Combinar la geometría con el factor de Boltzmann
La probabilidad real 
- La geometría: El número de formas en que se puede repartir esa energía (
- La termodinámica: La probabilidad de Boltzmann de que el baño térmico entregue esa energía (
).
Multiplicamos ambos factores:
Donde 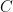
Paso 5: Calcular la constante de normalización (
Para que 
Para resolver esta integral, hacemos el cambio de variable clásico 


Sacamos las constantes 
La integral resultante es, por definición, la función Gamma de Euler, 

Paso 6: Obtención de la fórmula final
Sustituimos la constante
Reorganizando los términos de forma elegante para emparejar la energía con la energía térmica promedio (
🎨 Intuición física del resultado
Esta distribución (que matemáticamente es una distribución Gamma o Chi-cuadrado) nos muestra el triunfo de la estadística. Si tenemos por ejemplo un conjunto de moléculas orgánicas reaccionando con el oxígeno en un horno a temperatura T:
- A energías bajas, el término geométrico
frena la probabilidad porque hay «pocas formas» de repartir una energía tan pequeña entre tantos enlaces.
- A energías extremadamente altas, el factor de Boltzmann
- El pico de la curva ocurre exactamente en el valor dictado por el principio de equirrepartición:
, que es la energía interna promedio de las oscilaciones armónicas en las moléculas orgánicas.
Nota física: Si hacemos 


3. La nueva integral de la fracción eficaz f
Volviendo a la colisión reactiva, tenemos que hallar la fracción de moléculas que logran acumular una energía total igual o superior a la energía crítica
Para resolver esta integral, hacemos el cambio de variable 


Esta integral se resuelve por partes sucesivamente de forma iterativa (

Sustituyendo el valor original de 
Pasando a escala molar (
🧠 ¿Qué significa físicamente este cambio en la ecuación de Arrhenius?
Si unimos esta nueva fracción
Esto provoca dos consecuencias drásticas en la cinética de moléculas complejas como el azúcar o el almidón:
- La probabilidad de reacción aumenta masivamente: Como
, el factor multiplicativo
es un número enormemente grande. Esto compensa el factor estérico tan bajo (
–
) que, como comentamos anteriormente, tienen las moléculas orgánicas complejas. Significa que, aunque la orientación sea difícil, la molécula tiene «muchos bolsillos» (grados de libertad) donde guardar la energía del choque y redistribuirla internamente hasta que acaba alcanzando el enlace que puede romperse.
- Dependencia de la temperatura alterada: El factor preexponencial de Arrhenius (
. Al agrupar los términos, la constante de velocidad real se comporta como:
En la práctica experimental, debido a que el término exponencial 
Este almacenamiento de energía interna da pie a la teoría de Lindemann-Hinshelwood para reacciones unimolares, que explica cómo una molécula grande, tras chocar y absorber energía, puede tardar un tiempo en romperse de forma espontánea.
A nivel microscópico, los enlaces químicos no están aislados, sino que se comportan como resortes interconectados [1] que pueden transferir energía de unos a otros. Esta transferencia de energía se produce mediante dos mecanismos:
1. El resorte compartido (Acoplamiento cinético)
Los enlaces de una molécula comparten átomos comunes [1]. Si el enlace A (entre el átomo 1 y 2) vibra intensamente, el movimiento del átomo 2 arrastra inevitablemente al átomo 3, que forma el enlace B [1]. La masa del átomo intermedio actúa como un puente mecánico que transmite la fuerza física de la vibración [1].
2. La deformación de la nube electrónica (Acoplamiento potencial)
Cuando los átomos de un enlace se mueven, alteran la distribución de los electrones a su alrededor [2]. Como los electrones forman una «nube» continua en toda la molécula, el cambio de geometría en un extremo altera las fuerzas repulsivas y atractivas en el otro extremo [2]. Esta perturbación eléctrica empuja o tira de los enlaces vecinos [1, 2].
Si los enlaces fueran «resortes perfectos» (armónicos), la perturbación generada por el choque se propagaría de forma determinista por el sistema de múltiples resortes y al cabo de un tiempo (teorema KAM o Kolmogorov-Arnold-Moser de los sistemas dinámicos conservativos) volvería tan cerca como se quiera de la situación de partida. En realidad, al estirarse los enlaces se vuelven no-armónicos, esto es, responden con fuerzas de recuperación que no son estrictamente lineales. Ahora bien, como todo sistema no lineal de muchas interacciones, este sistema presenta “inestabilidad dinámica” o alta sensibilidad a las condiciones iniciales. Esto hace que colisiones aparentemente casi idénticas de dos moléculas orgánicas con una molécula del entorno provoquen en un caso la rotura rápida de algún enlace y en el otro caso ninguna rotura de enlace o una rotura tras un tiempo mucho más prolongado.
Pero tanto en el caso real no lineal como en el caso lineal (idealizado), el resultado es similar. Si la colisión ha introducido suficiente energía en algún punto de la molécula, tras un tiempo de propagación de las oscilaciones estas oscilaciones se pueden sumar en el lugar de un enlace frágil, y provocar su ruptura.
Aunque la mecánica estadística dicta que la energía se redistribuirá por toda la molécula a través de sus modos vibracionales, que algún enlace frágil de la molécula llegue a romperse depende de una feroz competencia de tiempos entre la redistribución intramolecular de la energía y los mecanismos de desactivación externa. La Teoría RRKM (Rice-Ramsperger-Kassel-Marcus) de reacciones unimolares y su dinámica molecular explica este fenómeno.
Cuando la molécula orgánica compleja recibe el impacto en una zona alejada de su enlace más frágil, esa energía en exceso entra inicialmente como energía vibracional localizada. A través de un fenómeno llamado Redistribución Intramolecular de Energía Vibracional (IVR), esa energía comienza a «viajar» e intercambiarse caóticamente entre todos los enlaces de la molécula. Este proceso es extremadamente rápido y suele completarse en escalas de tiempo de femtosegundos (

En un sistema real (fase líquida, disolución o gas a presión normal), la molécula compleja no está sin embargo aislada. Está rodeada de otras moléculas. El tiempo que tarda la energía en concentrarse estadísticamente «por azar» en el enlace frágil puede ser superior al tiempo que tarda la molécula en sufrir nuevos choques con el entorno (que se comporta como un baño térmico). Si otras moléculas con energía menor que la de activación la golpean antes, le «robarán» esa energía en exceso (desactivación colisional), enfriándola y evitando la rotura.
Contrariamente a la intuición, cuanto más grande y compleja sea la molécula orgánica, menor es la probabilidad de que el enlace frágil se rompa, incluso si el sistema está aislado. Una molécula compleja tiene decenas o cientos de grados de libertad vibracionales (modos de vibración). Al recibir el impacto, la energía se diluye entre todos esos modos. Para que el enlace frágil se rompa, las fluctuaciones estadísticas de la molécula deben lograr que una cantidad de energía superior a la de ruptura coincida simultáneamente en ese único enlace. Si la molécula es enorme, la probabilidad estadística de que la energía se concentre ahí de golpe es minúscula; la energía simplemente se queda almacenada como «calor interno» molecular.
Un escenario específico donde esa energía sí terminará concentrándose en el enlace débil y rompiéndolo es cuando la molécula está completamente aislada en el vacío o en fase gaseosa a muy baja presión. En este caso no puede transferir su calor a ninguna otra vecina. Al tener «tiempo infinito», la fluctuación estadística tarde o temprano concentrará la energía en el enlace frágil, provocando su ruptura.

Deja un comentario